SMODER Tutorial 02: Mouse Brain H3K27ac Result Visualization
This tutorial shows how to visualize representative SMODER outputs for the mouse brain RNA + H3K27ac peak example.
This tutorial continues from SMODER Tutorial 01: Mouse Brain H3K27ac Quick Start. In Tutorial 01, SMODER is run on the Mousebrain H3K27ac dataset and generates the main result file:
spatial_decon_result.h5ad
Here, we use this SMODER output file to generate downstream result visualizations.
What is spatial_decon_result.h5ad?
In SMODER, each dataset produces its own result file, conventionally named:
spatial_decon_result.h5ad
The same filename may appear in different output folders, but each file corresponds to a different dataset or run.
For the Mousebrain H3K27ac example, the file should contain:
adata.obsm["spatial"]
adata.obsm["cell_type_proportions"]
adata.obsm["embedding"]
adata.obsm["rna_encoder"]
adata.obsm["peak_encoder"]
These fields are used for spatial visualization, embedding-based clustering, and denoised signal reconstruction.
Load the SMODER result
from pathlib import Path
import scanpy as sc
spatial_result = Path("path/to/mousebrain_H3K27ac/spatial_decon_result.h5ad")
adata = sc.read_h5ad(spatial_result)
print(adata)
print(adata.obsm.keys())
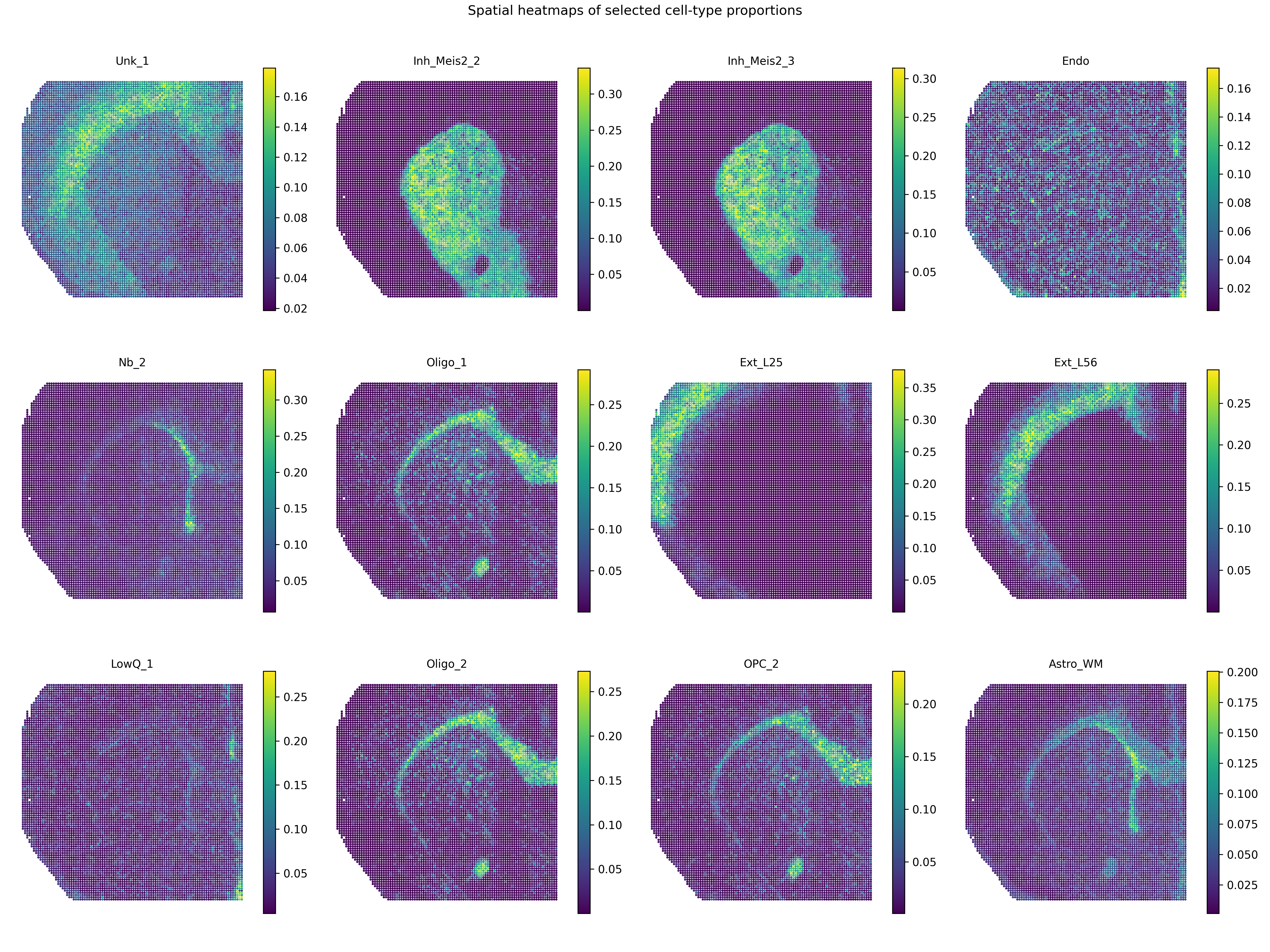
Plot cell-type proportion heatmaps
The inferred cell-type proportions are stored in:
adata.obsm["cell_type_proportions"]
The cell-type names are usually stored in adata.obs after metadata columns.
For the Mousebrain H3K27ac example, the first 9 columns of adata.obs are metadata, and cell-type names start from:
adata.obs.columns[9:]
A compact panel of cell-type proportion heatmaps can be generated by:
from smoder.visualization import plot_cell_type_proportion_panel
plot_cell_type_proportion_panel(
adata,
out_path="mousebrain_cell_type_proportion_top12.png",
obsm_key="cell_type_proportions",
obs_start_col=9,
top_n=12,
ncols=4,
title="Spatial heatmaps of selected cell-type proportions",
)
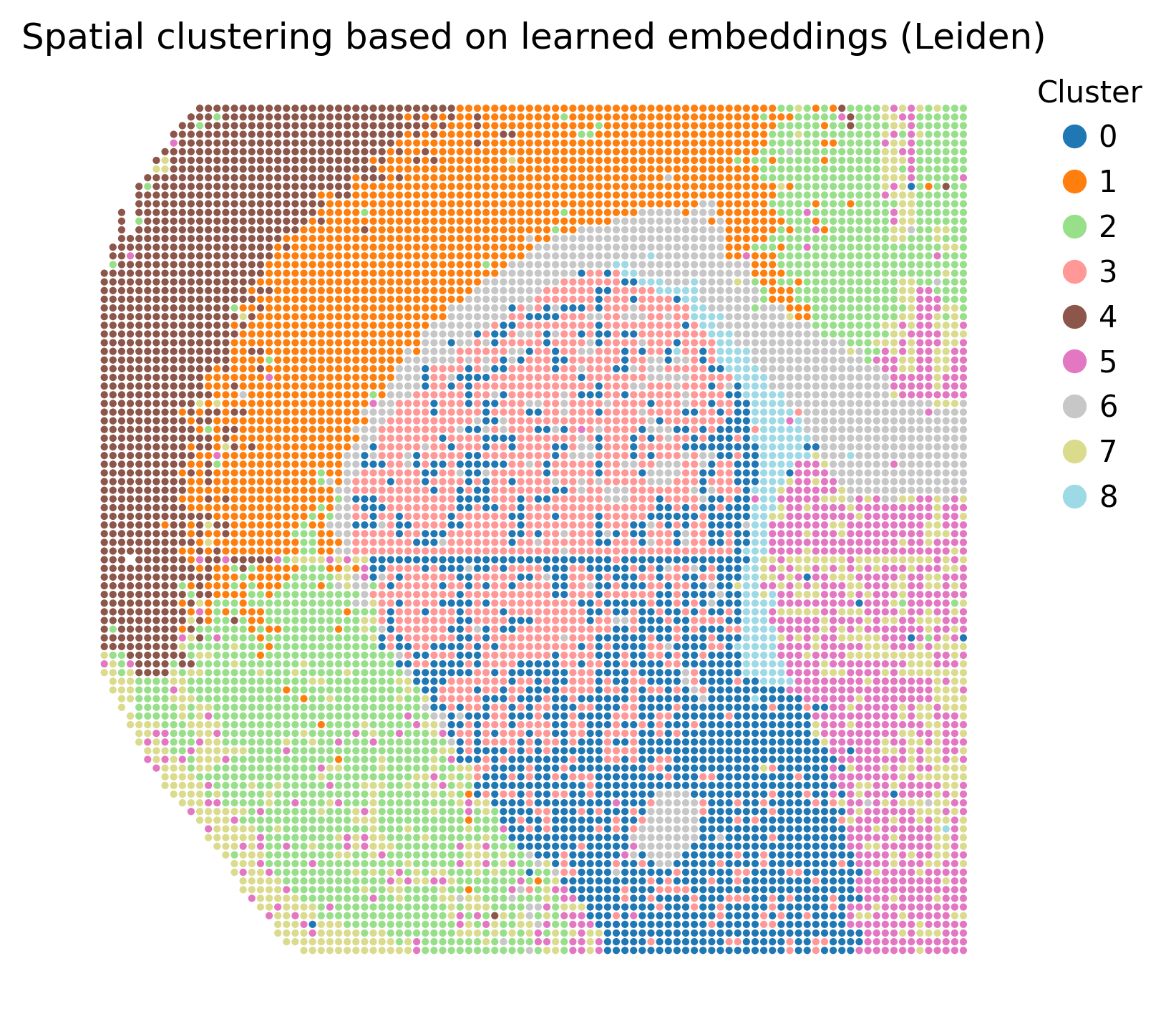
Plot spatial clusters from learned embeddings
The learned SMODER embedding is stored in:
adata.obsm["embedding"]
We can cluster this embedding and visualize the cluster labels spatially:
from smoder.visualization import plot_embedding_spatial_clustering
clustered = plot_embedding_spatial_clustering(
adata,
out_path="mousebrain_embedding_spatial_clustering.png",
embedding_key="embedding",
method="leiden",
resolution=0.6,
n_neighbors=15,
)
The resulting cluster labels are stored in:
clustered.obs["smoder_cluster"]
Reconstruct denoised RNA marker expression
SMODER stores RNA encoder representations in:
adata.obsm["rna_encoder"]
The function omics_reconstruct can reconstruct selected RNA marker genes from the trained SMODER representation.
from smoder.postprocessing import omics_reconstruct
rna_recon = omics_reconstruct(
omics_type="RNA",
expr_path="path/to/RNA.h5ad",
spatial_path="path/to/mousebrain_H3K27ac/spatial_decon_result.h5ad",
target_genes=["Penk", "Ppp1r1b", "Sez6l", "Gng2"],
encoder_key="rna_encoder",
hidden_dim=256,
n_layers=3,
epochs=500,
lr=1e-4,
patience=100,
save_path="RNA_recon.h5ad",
)
The reconstructed values are stored in rna_recon.X.
Then plot spatial heatmaps of the reconstructed RNA markers:
from smoder.visualization import plot_reconstruction_heatmaps
plot_reconstruction_heatmaps(
rna_recon,
out_dir="figures",
prefix="mousebrain_rna",
title_prefix="Denoised RNA expression",
)
Reconstruct denoised gene-level epigenomic signals
The second modality in this example is H3K27ac peak data. For gene-level denoised visualization, use a gene-level epigenomic target matrix and the SMODER peak encoder representation:
adata.obsm["peak_encoder"]
Run reconstruction with omics_type="EPIGENOMICS":
epi_recon = omics_reconstruct(
omics_type="EPIGENOMICS",
expr_path="path/to/gene_level_epigenomic_signal.h5ad",
spatial_path="path/to/mousebrain_H3K27ac/spatial_decon_result.h5ad",
target_genes=["Penk", "Ppp1r1b", "Sez6l", "Gng2"],
encoder_key="peak_encoder",
hidden_dim=256,
n_layers=3,
epochs=500,
lr=1e-4,
patience=100,
spatial_k=12,
do_preprocess=True,
save_path="epigenomics_recon.h5ad",
)
Then plot heatmaps of reconstructed gene-level epigenomic signals:
plot_reconstruction_heatmaps(
epi_recon,
out_dir="figures",
prefix="mousebrain_epigenomics",
title_prefix="Denoised gene-level epigenomic signal",
)
Complete plotting script
The repository also provides a plotting script that follows the same logic:
scripts/plot_mousebrain_h3k27ac_results_for_docs.py
In practice, you can adapt the path variables at the top of this script to your own local data locations.
Representative results
Cell-type proportion heatmaps
Spatial clustering from learned embeddings
Denoised RNA marker heatmaps
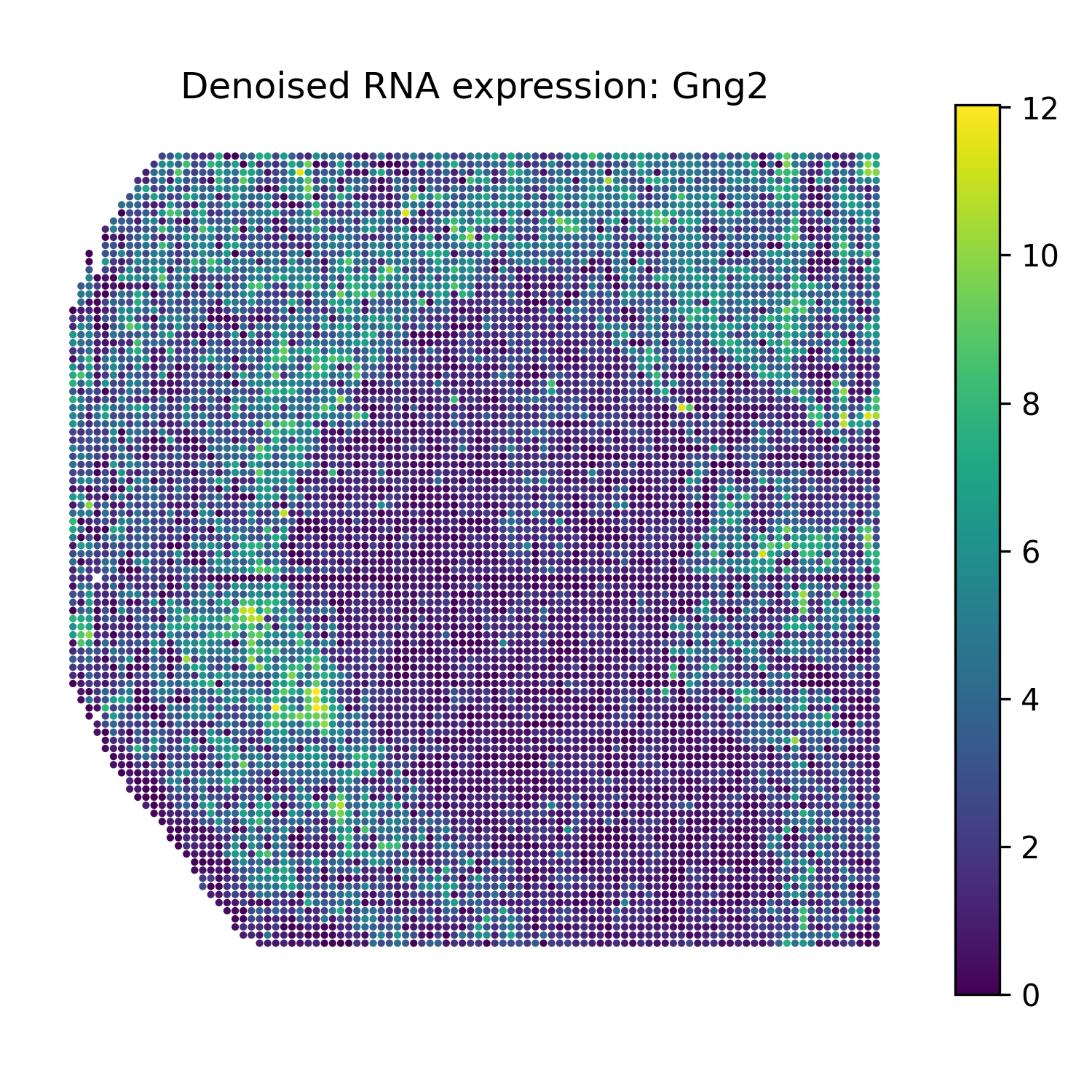
Gng2 |
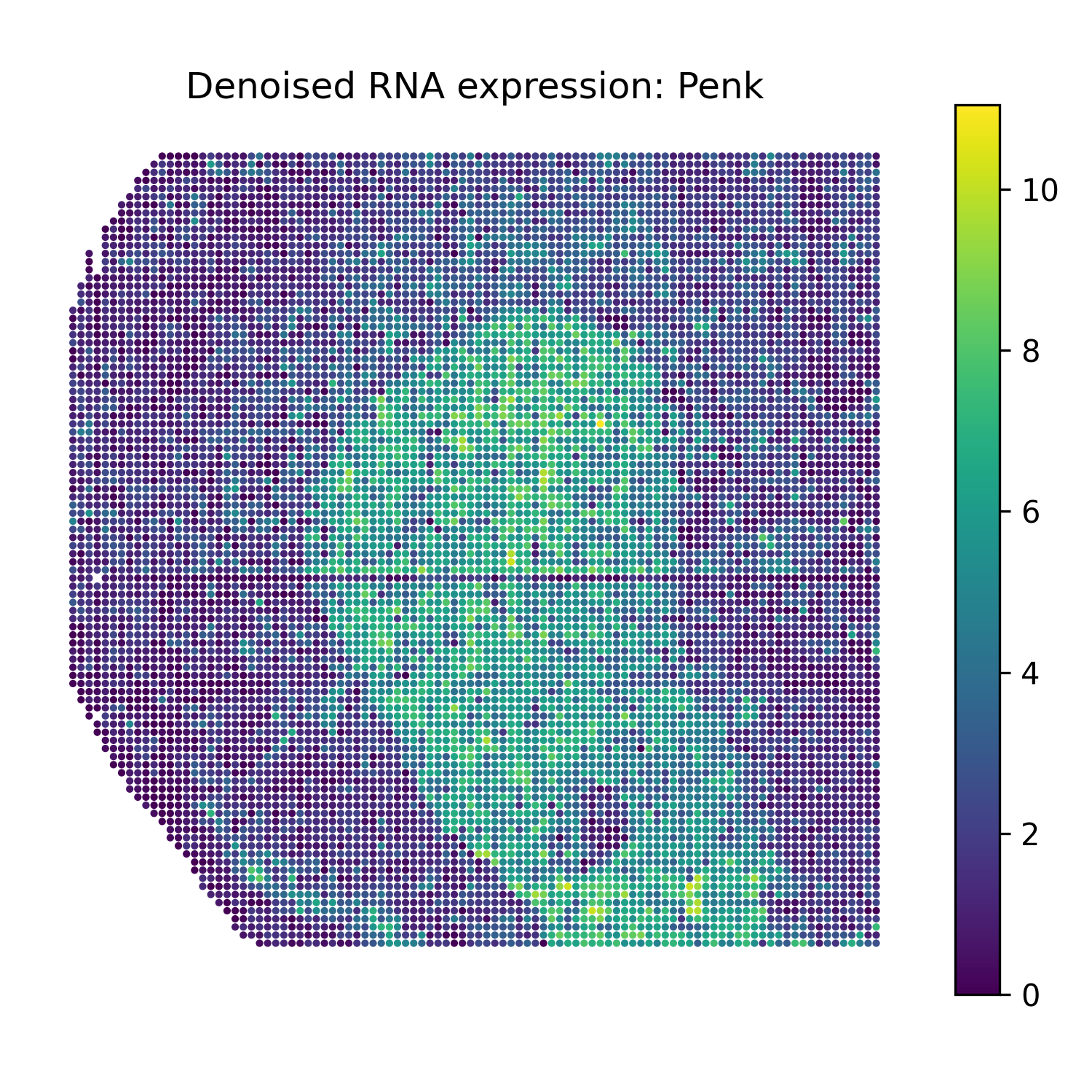
Penk |
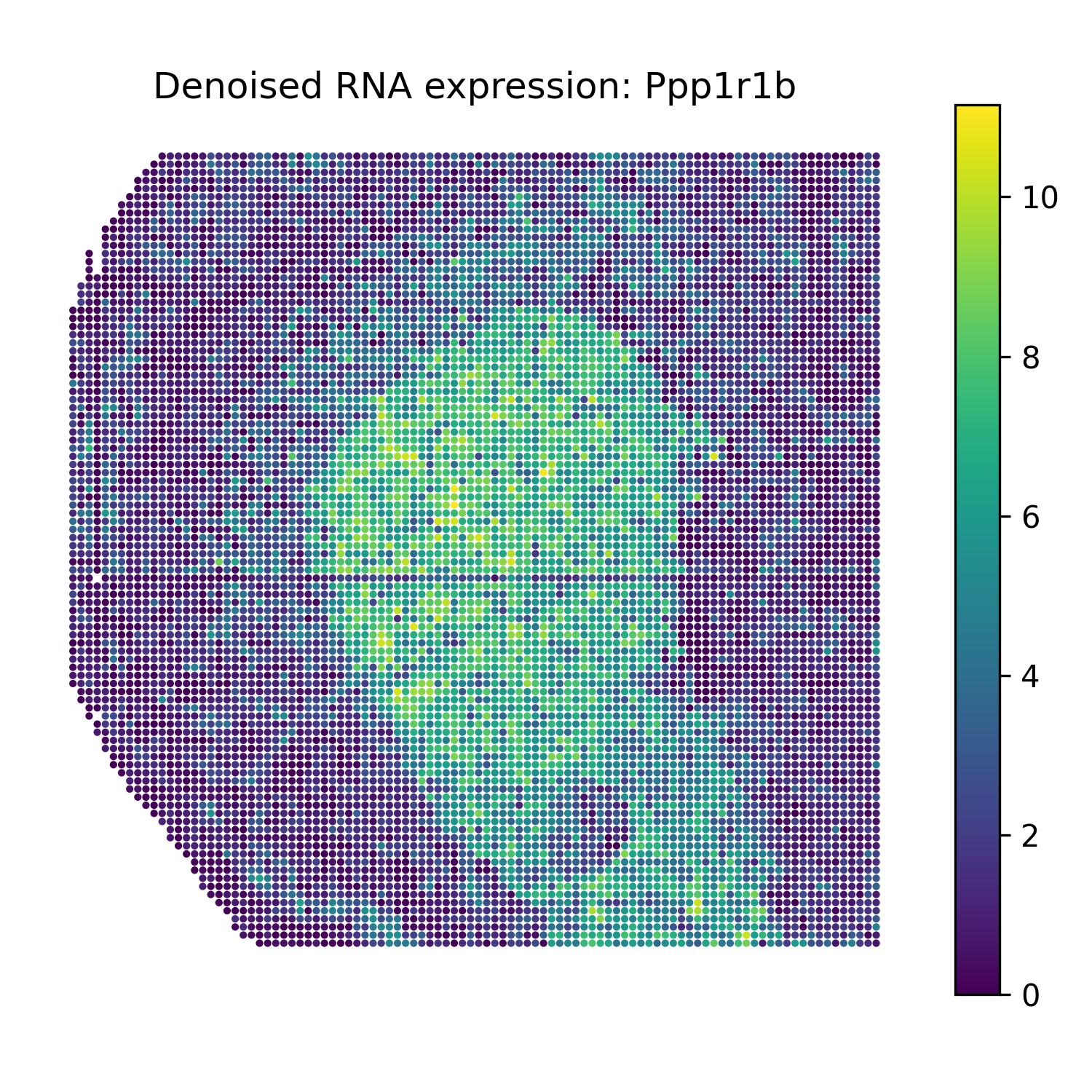
Ppp1r1b |
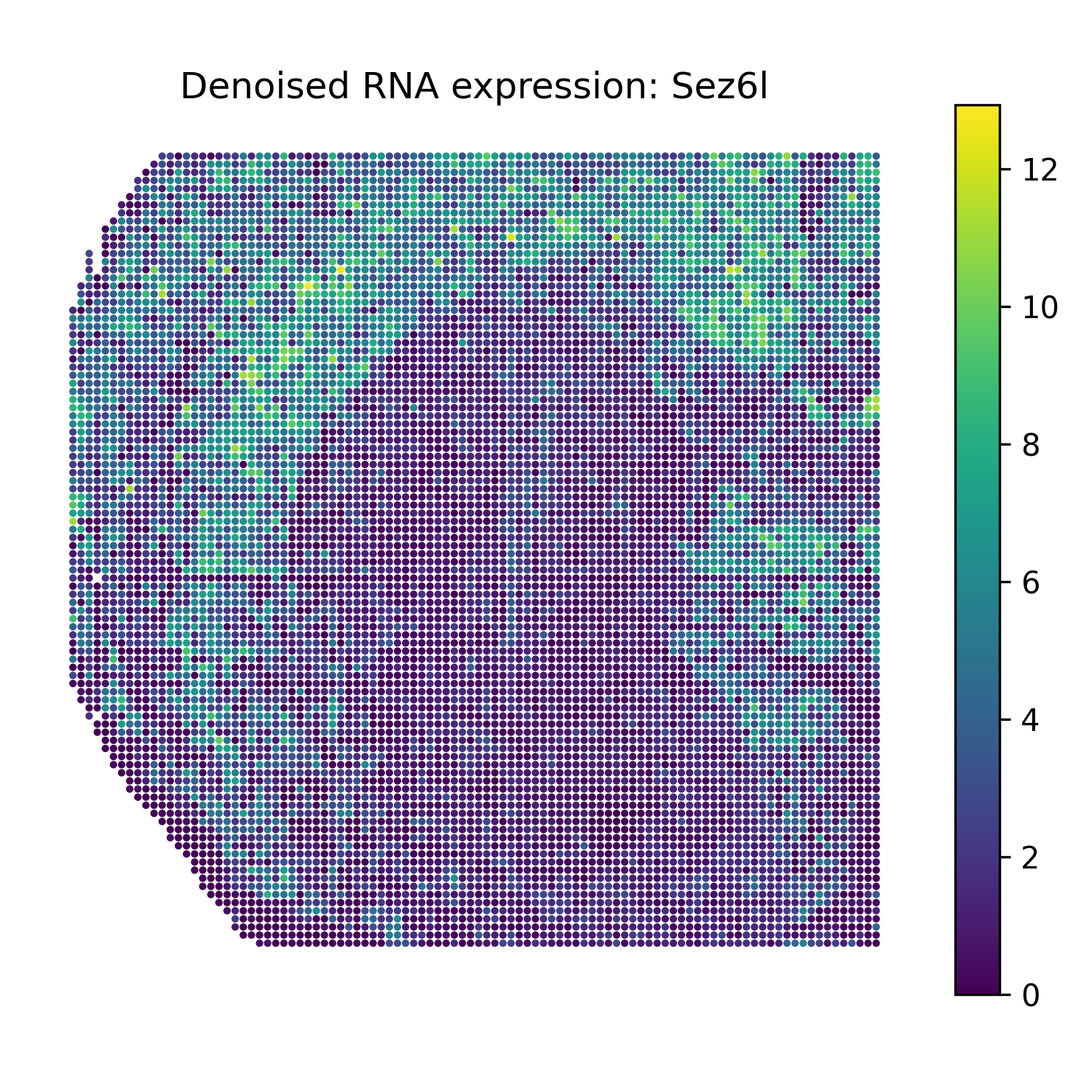
Sez6l |
Denoised gene-level epigenomic signal heatmaps
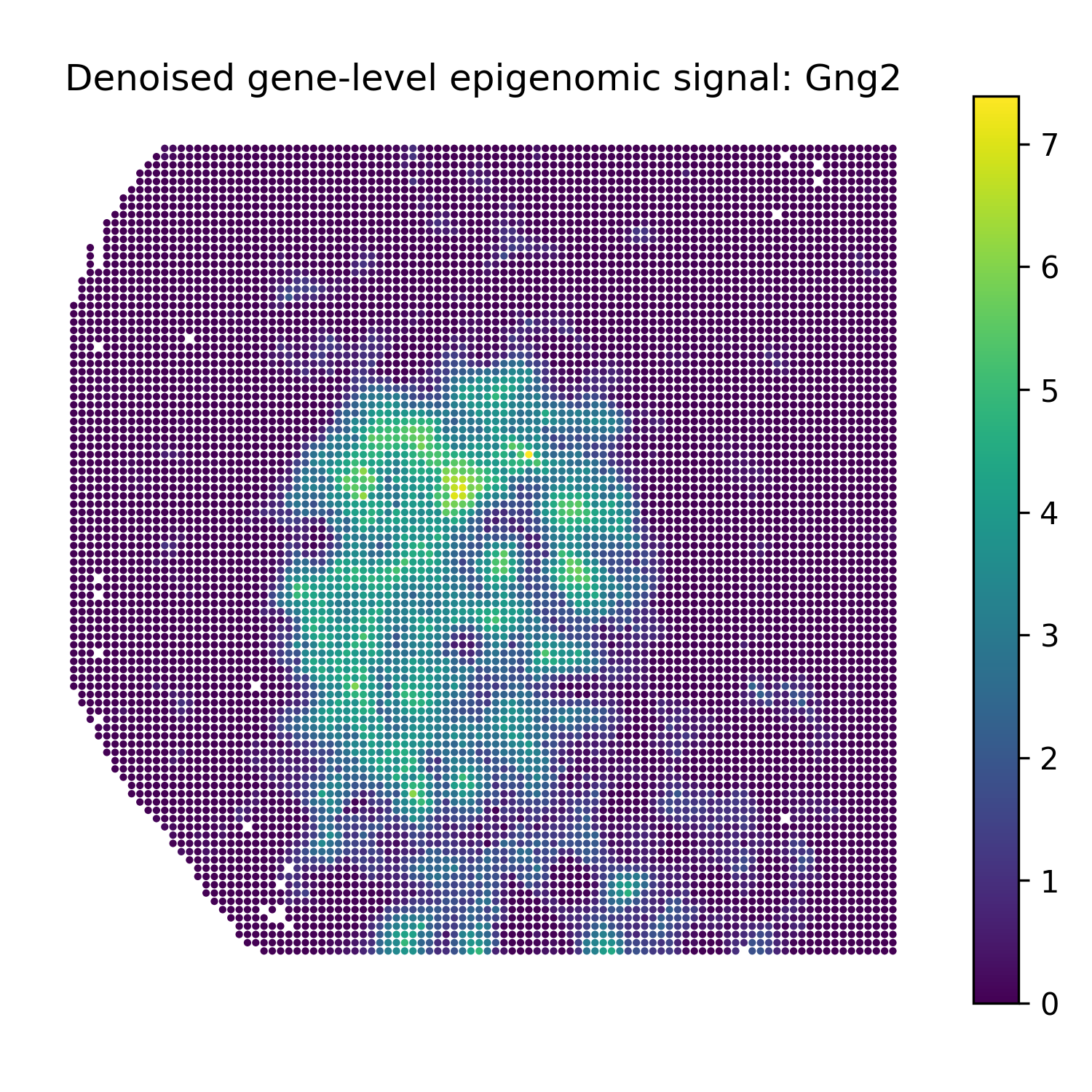
Gng2 |
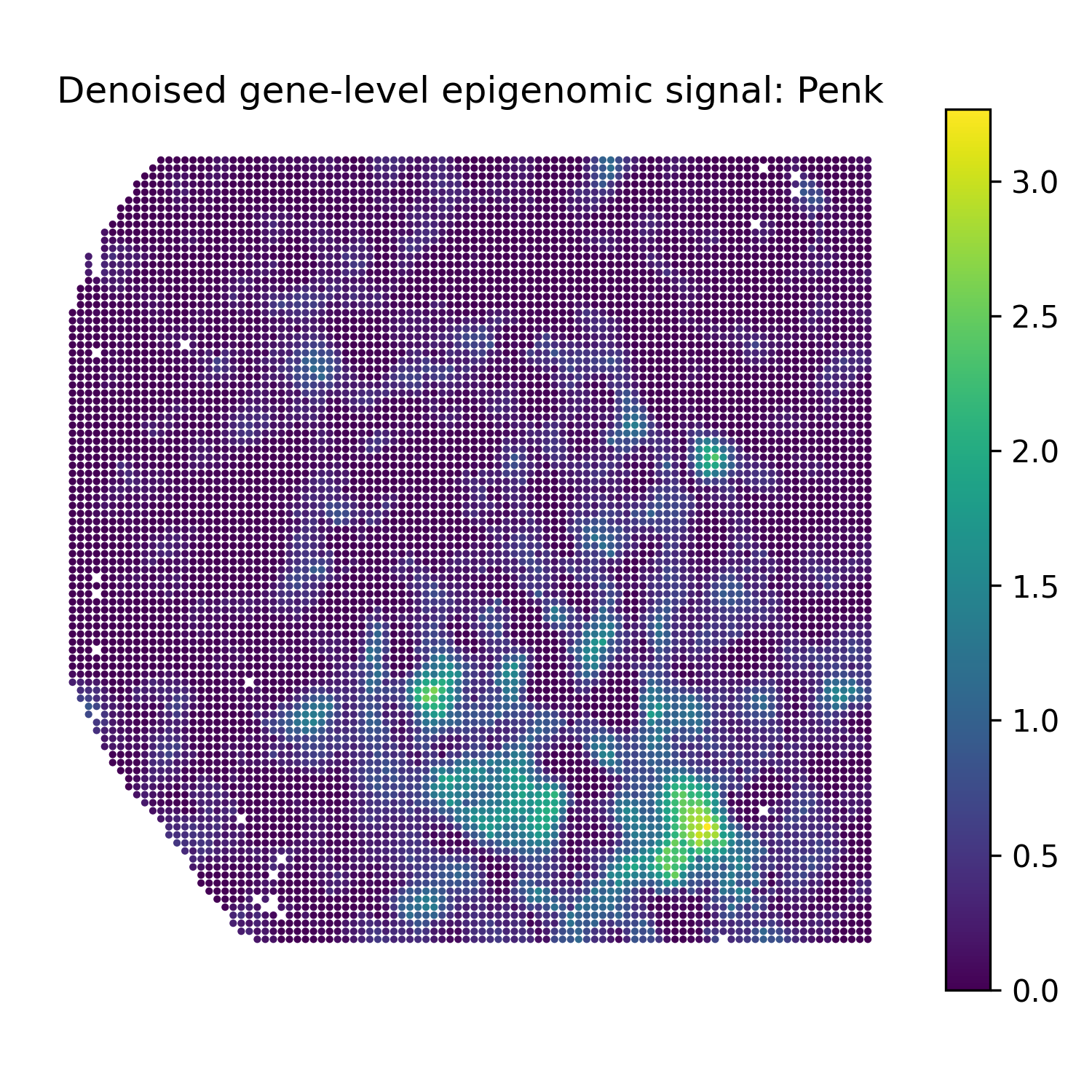
Penk |
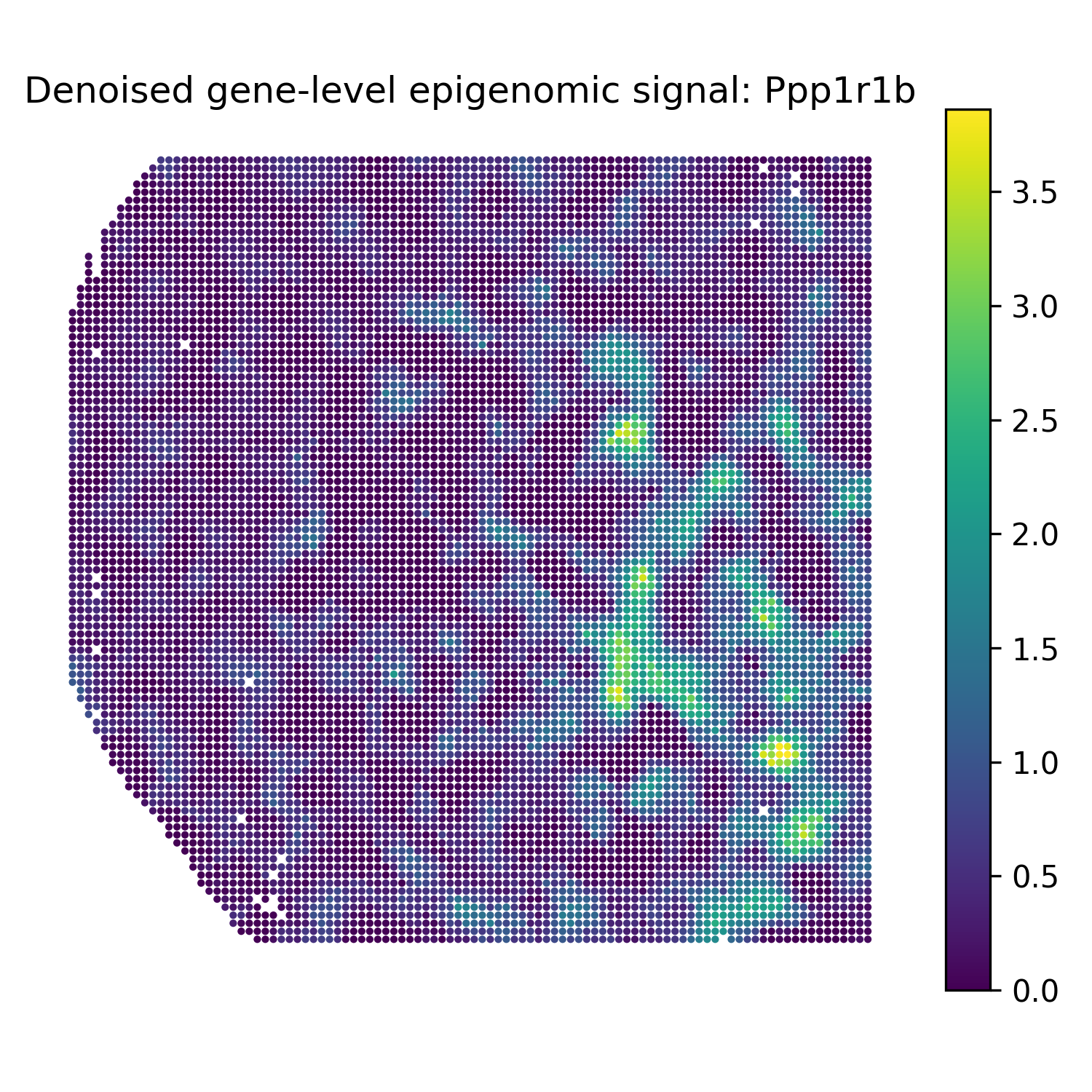
Ppp1r1b |
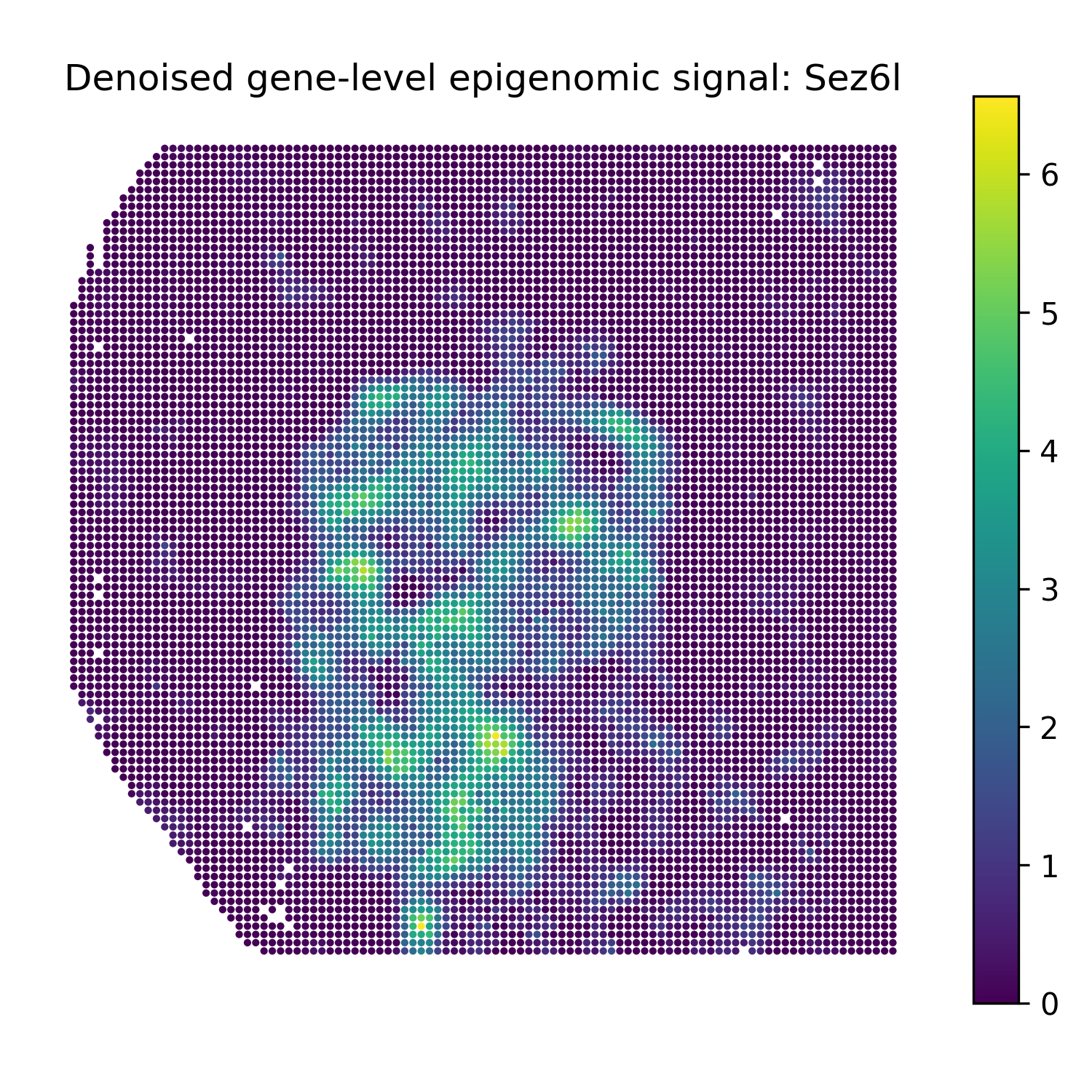
Sez6l |